Cookbook
Introduction
What is Jazzy?
Jazzy is a Python library that allows you to calculate a set of atomic/molecular descriptors which include the Gibbs free energy of hydration (kJ/mol), its polar/apolar components, and the hydrogen-bond strength of donor and acceptor atoms using either SMILES or MOL/SDF inputs. Jazzy is easy to use, does not require expensive hardware, and produces accurate estimations within milliseconds to seconds for drug-like molecules. The library also exposes functionalities to depict molecules with atomistic hydrogen-bond strengths in two or three dimensions.
What can I use Jazzy for?
The library was originally designed to support the processes of drug discovery and development but its applicability goes beyond life sciences. These are just some examples of the application of Jazzy:
To score compounds based on their molecular or atomic hydrogen-bond strengths or free energies of hydrations.
To describe molecules for machine learning purposes (e.g. physicochemical or ADME modelling).
To discriminate compounds on their C-H and X-H hydrogen-bond donor strengths.
To determine polar and apolar contributions of free energies of hydration.
What is this document for?
This document contains a comprehensive list of examples on how to use Jazzy. The library provides three levels of programmatic access which require increasing programming skills: (1) command-line interface (CLI), (2) API functions, and (3) core functions. This architecture aims to maximise accessibility, ease of integration, and compatibility across different versions of Jazzy - but it also encourages the development of new functionalities and the reutilisation of existing components. As a note for developers, Jazzy relies on RDKit and kallisto for the handling of molecule objects and the calculation of their atomic features.
Which functions should I use?
Depending on your programming expertise and what you are aiming to do with Jazzy, you should select an appropriate method to interact with it. Here, we are summarising what each level provides:
Command-line Interface: Terminal commands to predict properties against an individual SMILES string or a file containing a set of molecules. The CLI is the easiest way to use Jazzy and ensures the highest compatibility across versions. This is the level you might want to use if you are, for example, interested in just running Jazzy against a data set of molecules.
APIs: Python functions that are simple to use directly or to integrate within scripts or other software. These methods ensure high compatibility across versions and high performance without the need to implement the calculation logic from scratch. This is how you might want to access Jazzy, for example, if you are implementing a script where molecules are described before feeding a machine learning regressor.
Core Functions: Python functions that allow fine-grained configuration of the parameters used to calculate the descriptors and direct control on RDKit and kallisto. These methods are not necessarily cross-compatible in different versions of Jazzy, and they need to be integrated using appropriate exception handling. This might how you want to use Jazzy if you are planning to use only some of its functionalities or if you have already your chemoinformatics methods in place.
Feedback and Contributions
If you want to include new examples or review the existing ones, please refer to the Contributor Guide or submit a request to e.caldeweyher@gmail.com or ghiandoni.g@gmail.com.
Examples
Command-line Interface
Molecular Descriptors from SMILES
Please note that the CLI functionalities are beta and will be subjected to changes in the future.
Example of calculation of Jazzy descriptors for an individual SMILES string from the command line. Features include C-H donor strength (sdc), X-H donor strength (sdx) where X includes any non-carbon atoms, acceptor strength (sda), apolar contribution to delta g of hydration (dga), polar contribution to delta g of hydration (dgp), total delta G of hydration (dgtot) which also accounts for an interaction term that is not included in the results.
$ jazzy vec --opt MMFF94 'NC1=CC=C(C=C1)O'
{'sdc': 2.2437, 'sdx': 2.111, 'sa': 1.999, 'dga': -3.4321, 'dgp': -39.6424, 'tot': -43.0745, 'status': 'success', 'smiles': 'NC1=CC=C(C=C1)O'}
Atomistic Strength Visualisation from SMILES
Please note that the CLI functionalities are beta and will be subjected to changes in the future.
Example of calculation of atomic hydrogen-bond strengths for an individual SMILES string from the command line. Creates an SVG string of the molecule with its atomistic hydrogen-bond donor and acceptor strengths within a dictionary from an input SMILES string.
$ jazzy vis --opt MMFF94 'NC1=CC=C(C=C1)O'
{'svg': "<?xml version='1.0' encoding='iso-8859-1'?>\n<svg version='1.1' baseProfile='full'\nxmlns='http://www.w3.org/2000/svg'\nxmlns:rdkit='http://www.rdkit.org/xml'\nxmlns:xlink='http://www.w3.org/1999/xlink'\nxml:space='preserve'\nwidth='500px' height='500px' viewBox='0 0 500 500'>\n<!-- END OF HEADER -->\n<rect style='opacity:1.0;fill:#FFFFFF;stroke:none' width='500.0' height='500.0' x='0.0' y='0.0'> </rect>\n<path class='bond-0 atom-0 atom-1' d='M 406.4,222.5 L 369.6,230.9' style='fill:none;fill-rule:evenodd;stroke:#0000FF;stroke-width:2.0px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />\n<path class='bond-0 atom-0 atom-1' d='M 369.6,230.9 L 332.8,239.2' style='fill:none;fill-rule:evenodd;stroke:#000000;stroke-width:2.0px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />\n...</svg>\n", 'smiles': 'NC1=CC=C(C=C1)O', 'status': 'success'}
APIs
Molecular Features
Creates a dictionary of molecular features from a SMILES string. Features include C-H donor strength (sdc), X-H donor strength (sdx) where X includes any non-carbon atoms, acceptor strength (sda), apolar contribution to delta g of hydration (dga), polar contribution to delta g of hydration (dgp), total delta G of hydration (dgtot) which also accounts for an interaction term that is not included in the dictionary. Note that, if the SMILES cannot be processed, the function raises a JazzyError.
from jazzy.api import molecular_vector_from_smiles
molecular_vector_from_smiles("CC(=O)NC1=CC=C(C=C1)O")
{'sdc': 4.2822,
'sdx': 1.3955,
'sa': 2.461,
'dga': -3.0161,
'dgp': -51.2688,
'dgtot': -54.5831}
The molecular donor strength can simply be produced by summing C-H donor strength (sdc) and X-H donor strength (sdx):
from jazzy.api import molecular_vector_from_smiles
mol_vector = molecular_vector_from_smiles("CC(=O)NC1=CC=C(C=C1)O")
mol_vector["sdx"] + mol_vector["sdc"]
5.7138
Gibbs Free Energy of Hydration
Calculates the Gibbs free energy of hydration (kJ/mol) from a SMILES string. If the SMILES cannot be processed, the function raises a JazzyError.
from jazzy.api import deltag_from_smiles
deltag_from_smiles("CC(=O)NC1=CC=C(C=C1)O")
-54.5831
Atomic Features
Creates a list (of tuples) of tuples of atomic features from a SMILES string. Features include atomic number (z), formal charge (q), partial charge (eeq), atomic-charge dependent dynamic atomic polarizabilities (alp), hybridisation (hyb), number of lone pairs (num_lp), C-H donor strength (sdc), X-H donor strength (sdx) where X includes any non-carbon atoms, acceptor strength (sda). Note that, if the SMILES cannot be processed, the function raises a JazzyError.
from jazzy.api import atomic_tuples_from_smiles
atomic_tuples_from_smiles("[H]O[H]", minimisation_method="MMFF94")
[(('z', 8),
('q', 0),
('eeq', -0.6172),
('alp', 6.8174),
('hyb', 'sp3'),
('num_lp', 2),
('sdc', 0.0),
('sdx', 0.0),
('sa', 1.0)),
...
(('z', 1),
('q', 0),
('eeq', 0.3086),
('alp', 1.3102),
('hyb', 'unspecified'),
('num_lp', 0),
('sdc', 0.0),
('sdx', 1.0),
('sa', 0.0))]
The APIs also include atomic_map_from_smiles which is analoguous to atomic_tuples_from_smiles yet it produces its output as a list of dictionaries:
from jazzy.api import atomic_map_from_smiles
atomic_map_from_smiles("[H]O[H]", minimisation_method="MMFF94")
[{'z': 8,
'q': 0,
'eeq': -0.6172,
'alp': 6.8174,
'hyb': 'sp3',
'num_lp': 2,
'sdc': 0.0,
'sdx': 0.0,
'sa': 1.0,
'idx': 0},
...
{'z': 1,
'q': 0,
'eeq': 0.3086,
'alp': 1.3102,
'hyb': 'unspecified',
'num_lp': 0,
'sdc': 0.0,
'sdx': 1.0,
'sa': 0.0,
'idx': 2}]
Hydrogen-bond Strength Depiction
Creates an SVG rendering of the molecule with its atomistic hydrogen-bond donor and acceptor strengths from an input SMILES string. Note that, if the SMILES cannot be processed, the function raises a JazzyError. The depiction function accepts parameters to:
Create a two- or three-dimensional depiction (e.g.
flatten_molecule=Truegenerates a 2D molecule)Exclude specified types of strengths (e.g.
ignore_sa=Trueexcludes acceptor strengths from the rendering)Apply minimum strength thresholds (e.g.
sdc_threshold=0.7depicts sdc strengths only if greater than 0.7)Configure the rounding digits on the image (e.g.
rounding_digits=2rounds strengths to two digits)Configure the output size (e.g.
fig_size=[350,350]generates an image of 350x350 pixels)Depict strengths without highlighting their atoms (e.g.
highlight_atoms=False)Encode the image into base64 format (e.g.
encode=True)
from IPython.display import SVG
from jazzy.api import atomic_strength_vis_from_smiles
SVG(atomic_strength_vis_from_smiles(smiles="CC(=O)NC1=CC=C(C=C1)O",
flatten_molecule=True,
highlight_atoms=True,
ignore_sdc=False,
ignore_sdx=False,
ignore_sa=False,
sdc_threshold=0.7,
sdx_threshold=0.6,
sa_threshold=0.7,
rounding_digits=2))
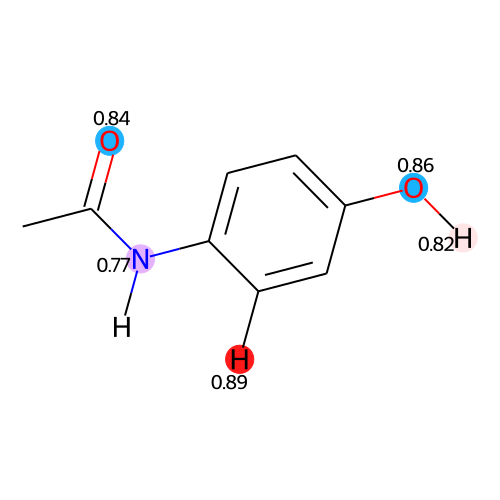
If you wish to convert an SVG image into PNG and save it within your machine, you can couple Jazzy with a library such as CairoSVG as follows:
from IPython.display import SVG
from cairosvg import svg2png
from jazzy.api import atomic_strength_vis_from_smiles
svg2png(bytestring=(atomic_strength_vis_from_smiles(smiles="CC(=O)NC1=CC=C(C=C1)O",
flatten_molecule=True,
highlight_atoms=True,
rounding_digits=2)),
write_to='output.png')
Core Functions
Jazzy calculates its descriptors using partial charges from kallisto and produces results that match the atomic indices generated by RDKit. This logic was conceived with the aim to facilitate the use and integration of Jazzy. This section reports some examples of how to use the core functionalities of the library.
RDKit and kallisto Molecules
To run Jazzy from the core, you need to produce both an RDKit and kallisto molecules. The kallisto molecule is used to produce the descriptors, and the RDKit molecule is where the rest of the chemoinformatics happens. The easiest way to produce a valid RDKit molecule to use rdkit_molecule_from_smiles with a SMILES string as an input. The function creates an RDKit object, adds explicit hydrogens to it, generates an embedding, and optionally runs an energy minimisation method against it. Note that, if the SMILES cannot be processed, the function returns a None.
from jazzy.core import rdkit_molecule_from_smiles
rdkit_molecule_from_smiles("NC1=CC=C(C=C1)O", minimisation_method="MMFF94")
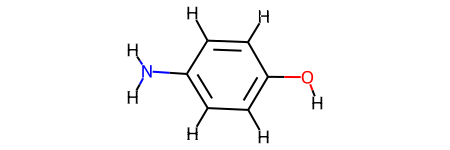
Alternatively, if you want to use an MDL molfile, you can construct a similar logic directly with RDKit:
import rdkit
rdkit_mol = rdkit.Chem.MolFromMolFile("../4_aminophenol.mol") # Creates the RDKit object
rdkit_mol = rdkit.Chem.AddHs(rdkit_mol, addCoords=True) # Adds hydrogens and their coordinates
rdkit_mol.__module__
'rdkit.Chem.rdchem'
Once you have an embedded RDKit molecule with explicit hydrogens, you can create its corresponding kallisto molecule:
from jazzy.core import rdkit_molecule_from_smiles, kallisto_molecule_from_rdkit_molecule
rdkit_mol = rdkit_molecule_from_smiles("NC1=CC=C(C=C1)O", minimisation_method="MMFF94")
kallisto_molecule_from_rdkit_molecule(rdkit_mol)
<kallisto.molecule.Molecule at 0x7f5e77c94340>
From Charges to Free Energy of Hydration
Given that you have both RDKit and kallisto molecules, a set of functions can be called sequentially to produce partial charges, atomic polar strengths, and free energy of hydration components.
Partial Charges
Jazzy wraps kallisto and maps its partial charges onto RDKit - i.e., the indices of the list of charges correspond to the atom indices in RDKit. This way, you can easily get results for your RDKit objects and carry on with your chemoinformatics logic.
from jazzy.core import rdkit_molecule_from_smiles, kallisto_molecule_from_rdkit_molecule, get_charges_from_kallisto_molecule
rdkit_mol = rdkit_molecule_from_smiles("NC1=CC=C(C=C1)O", minimisation_method="MMFF94")
kallisto_mol = kallisto_molecule_from_rdkit_molecule(rdkit_mol)
get_charges_from_kallisto_molecule(kallisto_mol, charge=0)
[-0.6627001925559142, 0.11268080360767647, -0.0873075526419112, -0.10025584673427702, 0.11201856315343292, -0.07343308047161486, -0.08901274832446447, -0.4865928417376737, 0.26238853128287026, 0.2622190860687737, 0.11949194864178196, 0.11522856894442372, 0.13396962850629238, 0.11992126184542574, 0.2613838704151783]
Atomic Strengths
The same principle described above applies to the generation of atomistic features: Jazzy creates a dictionary where keys are atom indices that match those in the RDKit molecule and values are dictionaries of features. Features include atomic number (z), formal charge (q), partial charge (eeq), atomic-charge dependent dynamic atomic polarizabilities (alp), hybridisation (hyb), number of lone pairs (num_lp), C-H donor strength (sdc), X-H donor strength (sdx) where X includes any non-carbon atoms, acceptor strength (sda).
from jazzy.core import rdkit_molecule_from_smiles, kallisto_molecule_from_rdkit_molecule
from jazzy.core import get_covalent_atom_idxs, get_charges_from_kallisto_molecule, calculate_polar_strength_map
rdkit_mol = rdkit_molecule_from_smiles("NC1=CC=C(C=C1)O", minimisation_method="MMFF94")
kallisto_mol = kallisto_molecule_from_rdkit_molecule(rdkit_mol)
atoms_and_nbrs = get_covalent_atom_idxs(rdkit_mol)
kallisto_charges = get_charges_from_kallisto_molecule(kallisto_mol, charge=0)
calculate_polar_strength_map(rdkit_mol, kallisto_mol, atoms_and_nbrs, kallisto_charges)
{
0: {'z': 7,
'q': 0,
'eeq': -0.6627,
'alp': 9.026,
'hyb': 'sp2',
'num_lp': 1,
'sdc': 0.0,
'sdx': 0.0,
'sa': 1.1157},
1: {'z': 6,
'q': 0,
'eeq': 0.1127,
'alp': 8.469,
...
'hyb': 'unspecified',
'num_lp': 0,
'sdc': 0,
'sdx': 0.5973,
'sa': 0}
}
Molecular Strengths
Atomic strengths are simply summed up to yield molecular strengths. Jazzy implements sum_atomic_map() within its helpers that does the job for you.
from jazzy.core import rdkit_molecule_from_smiles, kallisto_molecule_from_rdkit_molecule
from jazzy.core import get_covalent_atom_idxs, get_charges_from_kallisto_molecule, calculate_polar_strength_map
from jazzy.helpers import sum_atomic_map
rdkit_mol = rdkit_molecule_from_smiles("NC1=CC=C(C=C1)O", minimisation_method="MMFF94")
kallisto_mol = kallisto_molecule_from_rdkit_molecule(rdkit_mol)
atoms_and_nbrs = get_covalent_atom_idxs(rdkit_mol)
kallisto_charges = get_charges_from_kallisto_molecule(kallisto_mol, charge=0)
atomic_map = calculate_polar_strength_map(rdkit_mol, kallisto_mol, atoms_and_nbrs, kallisto_charges)
sum_atomic_map(atomic_map)
{'sdc': 2.2437, 'sdx': 2.111, 'sa': 1.999}
Free Energy of Hydration
The calculation of the free energy of hydration requires: RDKit molecule, kallisto molecule, the atomic hydrogen bonding strengths map, and a set of free parameters that are specific to each free energy component. Jazzy comes already with a a set of parameters. that we have derived by fitting the components against a set of experimental free energy of hydration values but you can replace them with your own parameters if you wish. Here we show an full example of how to calculate the delta hydration energy and its components for a SMILES string using the core functions.
If you are just interested in calculating the free energy of hydration without caring about the free parameters, we strongly advise to use directly the Free Energy of Hydration API.
# First of all, set the parameters
g0=1.884
gs=0.0467
gr=-3.643
gpi1=-1.602
gpi2=-1.174
gd=-0.908
ga=-16.131
expd=0.50
expa=0.34
gi=4.9996
f=0.514
from jazzy.core import rdkit_molecule_from_smiles, kallisto_molecule_from_rdkit_molecule
from jazzy.core import get_covalent_atom_idxs, get_charges_from_kallisto_molecule, calculate_polar_strength_map
from jazzy.core import calculate_delta_polar, calculate_delta_apolar, calculate_delta_interaction
# Then, let's create the molecules and their atomic strengths
smiles = "NC1=CC=C(C=C1)O"
rdkit_mol = rdkit_molecule_from_smiles(smiles, minimisation_method="MMFF94")
kallisto_mol = kallisto_molecule_from_rdkit_molecule(rdkit_mol)
atoms_and_nbrs = get_covalent_atom_idxs(rdkit_mol)
kallisto_charges = get_charges_from_kallisto_molecule(kallisto_mol, charge=0)
atomic_map = calculate_polar_strength_map(rdkit_mol, kallisto_mol, atoms_and_nbrs, kallisto_charges)
# Calculate individual terms and finally produce their sum
dgp = calculate_delta_polar(atomic_map,
atoms_and_nbrs,
gd=gd,
ga=ga,
expd=expd,
expa=expa)
dga = calculate_delta_apolar(rdkit_mol,
atomic_map,
g0=g0,
gs=gs,
gr=gr,
gpi1=gpi1,
gpi2=gpi2)
dgi = calculate_delta_interaction(rdkit_mol,
atomic_map,
atoms_and_nbrs,
gi=gi,
expa=expa,
f=f)
print(dgp + dga + dgi) # The sum of the terms yields the Free Energy of Hydration (kJ/mol)
-43.074539262505496
Additional Functionalities
Jazzy now supports passing certain parameters as keyword arguments to control its logic granularly. You can refer to Usage to see which functions accept which keywords. Parameters include the following:
Embedding Type
embedding_type configures whether the input molecule is embedded in 2D or 3D. Note that 2D embedding is much faster and less prone to failures but less accurate (see notebooks/gerber_deltag_validation.ipynb)
from jazzy.api import molecular_vector_from_smiles
molecular_vector_from_smiles("CC(=O)NC1=CC=C(C=C1)O", embedding_type="2D")
Embedding Seed
embedding_seed controls the seed used to embed the input molecule by RDKit (referred to as randomSeed).
from jazzy.api import molecular_vector_from_smiles
molecular_vector_from_smiles("CC(=O)NC1=CC=C(C=C1)O", embedding_seed=89)
Embedding Max Iterations
embedding_max_iterations configures the maximum number of iterations/attempts done by RDKit to embed the input molecule (referred to as maxIterations or maxAttempts). This parameter can be used to force to make Jazzy succeed/fail fast, which can be very beneficial for featurisation in real time.
from jazzy.api import molecular_vector_from_smiles
molecular_vector_from_smiles("CC(=O)NC1=CC=C(C=C1)O", embedding_max_iterations=3)